
02
Jan
MNGIE is clinically diagnosable because of a distinctive tetrad of gastrointestinal dysmotility progressive external ophthalmoplegia demyelinating neuropathy and. MNGIE is clinically diagnosable because.
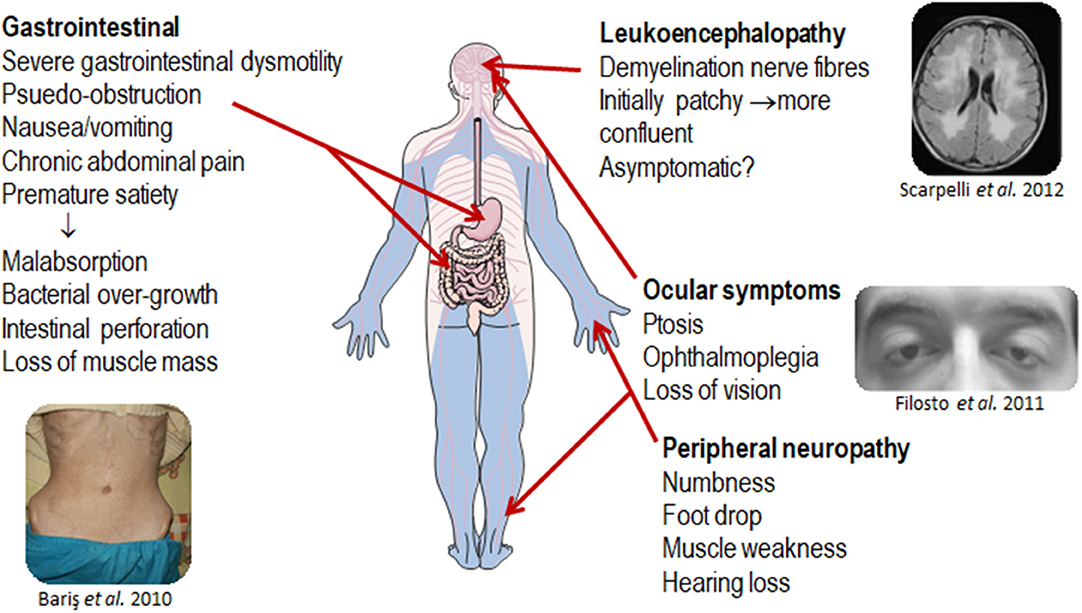
Mitochondrial neurogastrointestinal encephalopathy treatment. Physical therapy and occupational therapy may help preserve mobility. It is generally recommended that individuals with MNGIE syndrome avoid drugs that interfere with mitochondrial function including valproate phenytoin chloramphenicol tetracycline. Mitochondrial neurogastrointestinal encephalomyopathy MNGIE is a rare multisystem disorder characterized by progressive degeneration of the muscles of the gastrointestinal tract causing gastrointestinal dysmotility weakness of extra-ocular muscles causing drooping of the eyelids ptosis and restricted eye movements ophthalmoparesis degeneration of peripheral nerves causing altered.
Management is primarily supportive and includes attention to swallowing difficulties and airway protection. Dromperidone for nausea and vomiting. Gastrostomy and parenteral feeding for nutritional support.
Antibiotics for intestinal bacterial overgrowth. Amitriptyline nortriptyline and gabapentin for neuropathic symptoms. Objective To study the effect of continuous ambulatory peritoneal dialysis on nucleoside levels and clinical course in a patient with mitochondrial neurogastrointestinal encephalomyopathy MNGIE.
Patient We studied a patient with genetically verified MNGIE who prior to treatment had lost weight progressively developed amenorrhea vomited multiple times daily and had abdominal pain. Mitochondrial neurogastrointestinal encephalopathy MNGIE is a rare autosomal recessive condition. Deficiency of thymidine phosphorylase disrupts the nucleoside pool with progressive secondary mitochondrial DNA damage.
MNGIE is clinically diagnosable because. Mitochondrial neurogastrointestinal encephalomyopathy MNGIE is a fatal and rare autosomal recessive disorder of nucleotide metabolism caused by mutations in the nuclear thymidine phosphorylase gene TYMP which encodes cytosolic thymidine phosphorylase the enzyme required for the normal metabolism of pyrimidine deoxynucleosides thymidine and deoxyuridine. Mitochondrial neurogastrointestinal encephalomyopathy.
Approaches to diagnosis and treatment 1. Nishino I Spinazzola A Hirano M. Thymidine phosphorylase gene mutations in MNGIE a human mitochondrial disorder.
Hirano M Nishigaki Y Martí R. Mitochondrial neurogastrointestinal encephalomyopathy. Hematopoietic stem cell transplant hSCT is the best studied treatment modality and has been shown to provide a durable decrease in circulating nucleosides and sustained clinical improvement.
11 Although formal guidelines for hSCT are in place for MNGIE 12 the decision to proceed with treatment is complex because of the high-risk nature of the treatment and the debilitated state most patients are in by the. Treatment modalities that can increase thymidine phosphorylase activity and decrease thymidine and deoxy-uridine have shown symptomatic improvements in patients with MNGIE. Platelet transfusion hemodialysis peritoneal dialysis or allogeneic hematopoietic stem.
Mitochondrial neurogastrointestinal encephalopathy MNGIE is an autosomal recessive mitochondrial disorder caused by mutations in TYMP leading to reduced thymidine phosphorylase activity and high thymidine levels. Mitochondrial neurogastrointestinal encephalomyopathy treated with stem cell transplantation. A case report and review of literature.
Hematol Oncol Stem Cell Ther 2015885-90. Mitochondrial neurogastrointestinal encephalomyopathy MNGIE is a clinical syndrome that has been exclusively associated with autosomal recessive mutations in TYMP the gene encoding thymidine phosphorylase TPThe clinical hallmarks of MNGIE are ptosis ophthalmoparesis cachexia gastrointestinal dysmotility peripheral neuropathy and leukoencephalopathy evident on brain MRI. Mitochondrial neurogastrointestinal encephalopathy MNGIE disease is a condition that affects several parts of the body particularly the digestive system and nervous system.
The major features of MNGIE disease can appear anytime from infancy to adulthood but signs and symptoms most often begin by age 20. Mitochondrial neurogastrointestinal encephalopathy MNGIE is a rare autosomal recessive condition. Deficiency of thymidine phosphorylase disrupts the nucleoside pool with progressive secondary mitochondrial DNA damage.
MNGIE is clinically diagnosable because of a distinctive tetrad of gastrointestinal dysmotility progressive external ophthalmoplegia demyelinating neuropathy and. Mitochondrial neurogastrointestinal encephalomyopathy MNGIE is a rare autosomal recessive disorder characterized by severe muscle wasting gastrointestinal dysmotility leukoencephalopathy peripheral neuropathy and ophthalmoplegia. 1 The mutations in the ECGF1 gene encoding thymidine phosphorylase TP cause alterations in the respiratory chain and mitochondrial.
MitoPath is actively pursuing a gene therapy that uses a modified virus AAV28 to deliver millions of functioning copies of the TYMP gene to liver cells. We believe that this approach has a high likelihood of significantly improving the quality of life of patients with MNGIE. Mitochondrial neurogastrointestinal encephalomyopathy MNGIE is an autosomal recessive disease caused by mutations in the thymidine phosphorylase gene TYMP.
1 TYMP encodes for thymidine phosphorylase TP which catabolizes thymidine dThd and deoxyuridine dUrd into their respective bases. TYMP mutations markedly reduceabolish TP activity.
Previous post
Mixed bacteria in urineNext post
Mitochondrial dna damage assay